
PUBLICATIONS
At UC Merced

Bench-Stable Iron Bipyridine Complexes as Long-Lived Catalysts for the Hydrodefluorination of Arenes at Room Temperature
Harleen Kaur, Diksha Sharma, Rebeca Arevalo*
Preprint: ChemRXiv 2026, 10.26434/chemrxiv.15001775/v1
The iron(II) complex [Fe(tBubipy)OTf2] (tBubipy = 4,4’-ditertbutyl-2,2’-bipyridine, OTf = CF3SO3-) was found an efficient and bench-stable precatalyst for the hydrodefluorination (HDF) of arenes upon in situ activation with NaHBEt3 employing H2SiPh2 as H source at room temperature. The identity of the bipy ligand as well as of the X type ligands in the precatalyst were key to enable high efficiency for the process. Polyfluorinated arenes containing a different number of fluorine atoms in different positions were hydrodefluorinated to afford exclusive products from single HDF or mixtures of arenes from single and sequential HDF. The system showed a remarkable efficiency for 2,3,5,6-tetrafluoroanisole, for which a TON of 31.6 was obtained at room temperature. The efficiency of the system increased at high temperature, catalyzing up to 5 HDF cycles for hexafluorobenzene at 100 °C. The highest catalytic efficiency was found for tetrafluoroarenes, followed by tri- and perfluorinated arenes. A marked preference for fluorine atoms located between C–F and C–H bonds was observed in tetra- and trifluoroarenes, the efficiency of the system being primarily governed by the number of fluorine atoms in this environment, while their relative positions determined the efficiency when the number was identical. Mechanistic experiments for the hydrodefluorination of 2,3,5,6-tetrafluoroanisole suggested that paramagnetic Fe(II) are responsible for catalysis. The high catalytic efficiency of the system was attributed to the extended lifetime of the catalyst resting states, arising from the robust five-membered chelate formed by the tBubipy ligand, which prevented catalyst deactivation by ligand decoordination.

Triethoxysilane-Catalyzed Hydroboration of Alkenes, Nitriles, Ketones and Esters: From Non-Covalent Interactions to a Hydrosilylation/Si-B Exchange Pathway
Harleen Kaur, Diksha Sharma, Rebeca Arevalo*
Preprint: ChemRXiv 2026, 10.26434/chemrxiv.15000710/v1
Triethoxysilane was found an efficient precatalyst for the hydroboration of alkenes, nitriles, ketones and esters with pinacolborane (HBPin). The catalyst showed an excellent substrate scope including aromatic and aliphatic substrates affording the products in good to excellent yields. The system was found less efficient for the hydroboration of alkenes and Ealkenylboronate esters than that of alkynes, which could be traced back to the strength of the interactions between the multiple CC bonds of the substrates with HSi(OEt)3. The nature of the multiple CC bond (C=C vs C≡C) played the largest role in catalyst efficiency, the presence of a BPin substituent in the E-alkenylboronate esters not impacting the substrate conversion compared to that of terminal alkenes. The nature of aryl substituents in aromatic substrates exerted distinct effects on catalytic efficiency depending on the substrate class. Whereas the catalyst was sensitive to both the electronic and steric properties of terminal alkynes, catalytic efficiency in the hydroboration of aromatic nitriles was governed primarily by steric effects, with minimal sensitivity to electronic variation. Competition experiments allowed to unravel the catalyst preferences for the hydroboration of different types of functional groups, HSi(OEt)3 showing a significant preference for alkynes and alkenes over nitriles and ketones. Unlike in the HSi(OEt)3-catalyzed sequential hydroboration of alkynes, the fluorinated analog HSi(OCH2CF3)3 showed a comparable efficiency to HSi(OEt)3 for the hydroboration of nitriles, suggesting that the Lewis acidity at Si did not play a role in catalyst efficiency. The significant differences between alkynes and heteroatom-containing substrates could be traced back to the presence of a catalytically active species different from HSi(OEt)3 in the hydroboration of nitriles, ketones and esters. Additionally, intermediates from hydrosilylation of the multiple bond of nitriles were identified during catalytic turnover, supporting a mechanism involving H-Si bond addition across the multiple bond followed by Si-B exchange steps.

Iron-catalyzed Hydrodefluorination of Polyfluorinated Arenes: Wide Bite-Angle Phosphine Ligands Lead to High Defluorination Degree and Broad Substrate Scope
Diksha Sharma, Rebeca Arevalo*
Preprint: ChemRXiv 2025, 10.26434/chemrxiv-2025-4599v
The iron(II) complex [Fe(CyXantphos)Cl2] (CyXantphos = 4,5-bis(dicyclohexylphosphino)-9,9-dimethylxanthene) was found an efficient precatalyst for the hydrodefluorination of arenes employing HBPin and NaHBEt3 or NaOtBu as in situ activators. Polyfluorinated arenes containing a different number of fluorines in different positions were hydrodefluorinated to afford exclusive products from single HDF or mixtures of arenes from single and sequential HDF. This system afforded the highest yields and broader substrate scope reported to date for Fe-catalyzed HDF of arenes. The identity of the in situ activator (NaHBEt3 or NaOtBu) impacted the efficiency of the system resulting in higher degree of HDF and a higher efficiency for low fluorination degree arenes (1-3 F atoms) when NaOtBu was employed. The system tolerated the presence of other substituents (CF3, CH3, OCH3) at the aryl ring, with substitution of tetrafluoroarenes lead to para-regioselective HDF in all cases, with the identity of the substituent impacting catalyst efficiency. Mechanistic experiments for the hydrodefluorination of tetrafluoroanisole suggested that paramagnetic Fe(II) hydrides are responsible for C-F activation in both catalytic systems, with different identity depending on the activator employed. In contrast, diamagnetic iron(II) complexes were found to be catalytically relevant intermediates and the catalyst resting-states were found to be different depending on the activator identity, with the resting-states of the NaHBEt3 system being longer lived than those in the NaOtBu, consistent with the higher efficiency of NaHBEt3 in situ activation for this substrate.

Sustainable Catalysis for Borylation: Earth-Abundant Homogeneous Catalysts and Their Mechanisms
Rebeca Arevalo, Himani Ahuja,‡ Victor Duran Arroyo,‡ Harleen Kaur‡
ACS In Focus, DOI: 10.1021/acsinfocus.7e9018
‡ These authors contributed equally
This primer is intended for students beginning their research journey into the exciting world of homogeneous catalysis. We hope it inspires you to think creatively about catalyst design—often the first step toward unlocking transformations once believed to be out of reach. Whether you are new to the field or revisiting its fundamentals, we invite you to explore these concepts with curiosity and imagination.

Bis(Cyclooctadiene)Nickel(0)-Catalyzed Exhaustive C(sp2) and C(sp3)-X Hydrodehalogenation of Pyridines and Arenes Employing Ammonia Borane and Potassium Tert-Butoxide
Himani Ahuja, Rebeca Arevalo*
Inorg. Chem. Front. 2026, 13, 3079 - 3095, DOI: 10.1039/D6QI00010J
The hydrodefluorination (HDF) of CF3 groups and the hydrodehalogenation (HDH) of C(sp2)-X (X = F, Cl, Br) bonds in pyridines was conducted employing [Ni(COD)2] (COD = 1,5-cyclooctadiene) as catalyst and NH3BH3 as the H source in the presence of KOtBu at room temperature. The system was found efficient for the exhaustive hydrodefluorination of CF3 groups and of C(sp2)-X bonds at the 2, 3 or 4-positions of pyridines and for the selective HDH of C(sp2)-X bonds of trifluoromethylpyridines while retaining the CF3 group. Additionally, the system also showed a good efficiency for the C(sp2)-X (X = F, Cl, Br) HDH of halogenated pyridines without CF3 groups and arenes with low to moderate halogenation degree (1-4 halogens). Mechanistic studies allowed to identify three simultaneous HDF cycles operative in the HDF of 2-trifluoromethylpyridine with the first cycle being the rate-determining and the reaction of the [Ni(COD)2] precatalyst with the H source as the entry pathway to the cycle.


Nickel-Catalysed Sequential Hydrodefluorination of Pyridines: Mechanistic Insights Led to the Discovery of Bench-Stable Precatalysts
Victor Duran Arroyo, Roger Nunez, Rebeca Arevalo*
Inorg. Chem. Front. 2025, 12, 5303-5314
The nickel(0) complex [Ni(iPrPN)(COD)] (iPrPN = 2-[(N-diisopropylphosphino)methylamino]pyridine, COD = 1,5-cyclooctadiene) was an efficient precatalyst for the hydrodefluorination of pyridines employing pinacolborane (HBPin). 2-fluoro and 2,6-difluoropyridines were hydrodefluorinated at the 2- and 6-positions at room temperature in 3 h 30 min. The impact of the number of fluorine atoms and their position at the pyridyl ring in the efficiency of the catalyst was explored. Mechanistic experiments for the hydrodefluorination of 2,6-difluoropyridine allowed to identify COD decoordination followed by C-F oxidative addition as the catalyst entry pathway to the cycle and the [Ni(iPrPN)(COD)] complex as the catalyst resting-state. The Ni(II) fluoride complexes, [NiF(iPrPN)(6-Fpy)] (6-Fpy = 6-fluoropyrid-2-yl) and [NiF(iPrPN)(py)] (py = 2-pyridyl) were independently synthesized and identified as intermediates in the two subsequent hydrodefluorination cycles operative through single-turnover experiments. Both Ni(II) fluoride complexes were found to be bench-stable precatalysts for the process with a comparable efficiency to [Ni(iPrPN)(COD)] in the presence of a substoichiometric amount of COD to prevent catalyst deactivation.

Triethoxysilane-Catalyzed Sequential Regioselective Hydroboration of Terminal Alkynes: Sustainable Access to E-Alkenylboronate and Alkyl Gem-Diboronate Esters
Harleen Kaur, Himani Ahuja, Rebeca Arevalo*
ACS Catal. 2025, 15, 976-981
The commercial reagent, HSi(OEt)3, was found an efficient catalyst for the synthesis of E-alkenylboronate esters and alkyldiboronate esters by single or double hydroboration of terminal alkynes with HBPin (pinacolborane). The reaction time controlled whether a single or a double addition of HBPin to the alkynes occurred. Aromatic terminal alkynes containing strong electron-donating or -withdrawing substituents at different positions as well as aliphatic alkynes were efficiently mono- and dihydroborated. Mechanistic studies suggest that the formation of diboronate esters proceeds by a double hydroboration with the second hydroboration cycle being the rate-determining. The reaction of HSi(OEt)3 with HBPin has been identified as a catalyst deactivation pathway operative in catalysis. Screening of other silicon compounds for this transformation support that the presence of three OEt groups in HSi(OEt)3 is key to promote the second hydroboration step.

Tandem Manganese Catalysis for the Chemo-, Regio-, and Stereoselective Hydroboration of Terminal Alkynes: In Situ Precatalyst Activation as a Key to Enhanced Chemoselectivity
Victor Duran Arroyo, Rebeca Arevalo*,
RSC Adv. 2024, 14, 5514-5523
The manganese(II) complex [Mn(iPrPNP)Cl2] (iPrPNP = bis(diisopropylphosphine)pyridyl)) was found to catalyze the stereo- and regioselective hydroboration of terminal alkynes employing HBPin (pinacolborane). In the absence of in situ activators, mixtures of alkynylboronate and E-alkenylboronate esters were formed, whereas when NaHBEt3 was employed as in situ activator, E-alkenylboronate esters were exclusively accessed. Mechanistic studies revealed a tan-dem C-H borylation / semihydrogenation as the pathway accounting for the formation of the products. Stoichiometric reactions hint toward reaction of a Mn-H active species with the terminal alkyne as the catalyst entry pathway to the cycle, whereas reaction with HBPin led to catalyst deactivation.

Chemoselective C(sp)-H Borylation of Terminal Alkynes Catalyzed by a Bis(N-heterocyclicsilylene) Manganese Complex
Himani Ahuja,‡ Harleen Kaur,‡ Rebeca Arevalo*
Inorg. Chem. Front. 2023, DOI: 10.1039/D3QI01033C.
‡ These authors contributed equally
The manganese(II) complex [Mn(SiNSi)Cl2] (SiNSi = 2,6-[EtNSi(NtBu)2CPh]2C5H3N) was an efficient catalyst for the chemoselective C(sp)-H borylation of terminal alkynes. Aliphatic as well as aromatic alkynes containing electron-withdrawing and -donating substituents in different positions have been efficiently borylated. In all cases, the cata-lyst showed an excellent chemoselectivity towards C-H borylation and the reactions proceeded without additives or in-situ activators. Paramagnetic Mn complexes are involved in catalytic turnover which is proposed to occur by a re-dox-neutral Mn(II) cycle. Stoichiometric reactions support that the [Mn(SiNSi)Cl2] precatalyst enters the catalytic cycle by reaction with HBPin. KIE experiments point toward C-H activation of the alkyne as not being involved in the rate-determining step.

PUBLICATIONS
Prior to UC Merced

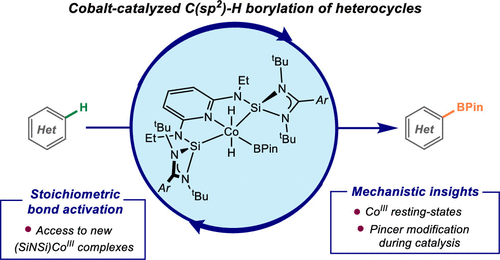
C(SP2)–H BORYLATION OF HETEROCYCLES BY WELL-DEFINED BIS(SILYLENE)PYRIDINE COBALT(III) PRECATALYSTS: PINCER MODIFICATION, C(SP2)–H ACTIVATION, AND CATALYTICALLY RELEVANT INTERMEDIATES
Rebeca Arevalo, Tyler P. Pabst, Paul J. Chirik
Organometallics 2020, 39, 2763–2773
RHENIUM-CATALYSED HYDROBORATION OF ALDEHYDES AND ALDIMINES
Rebeca Arevalo, Christopher M Vogels, Gregory A. MacNeil, Lucia Riera, Julio Perez, Stephen A. Westcott,
Dalton Trans. 2017, 46, 7750-7757

Rebeca Arevalo, Lucia Riera, Julio Perez
Inorg. Chem. 2017, 56, 4249-4252
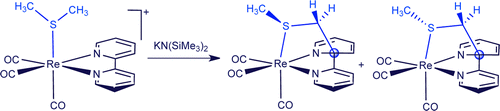
NUCLEOPHILIC ADDITIONS TO COORDINATED 1,10-PHENANTHROLINE: INTRAMOLECULAR, INTERMOLECULAR, REVERSIBLE, AND IRREVERSIBLE
Rebeca Arevalo, M. Isabel Menendez, Ramon Lopez, Isabel Merino, Lucia Riera, Julio Perez
Chem. Eur. J. 2016, 22, 17972-17975
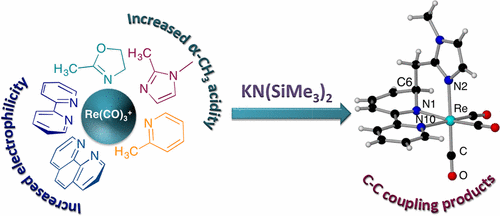
INTERLIGAND C-C COUPLING BETWEEN Α-METHYL N-HETEROCYCLES AND BIPY OR PHEN AT RHENIUM TRICARBONYL COMPLEXES

DEPROTONATION OF COORDINATED PHOSPHANES IN A RHENIUM COMPLEX: C-C COUPLING WITH DIIMINE COLIGANDS
Rebeca Arevalo, Julio Perez, Lucia Riera
Chem. Eur. J. 2015, 21, 3546-3549

BOOK CHAPTERS
Prior to UC Merced

DEAROMATIZATION OF TRANSITION METAL-COORDINATED N-HETEROCYCLIC LIGANDS AND RELATED CHEMISTRY
Rebeca Arévalo, Maialen Espinal-Viguri, Miguel A. Huertos, Julio Pérez and Lucía Riera
Advances in Organomet. Chem. 2016, 65, 47-106